




{kind=link}
Una investigación liderada por Antonio Salas y Federico Martinón, de la Facultade de Medicina de la USC y el IDIS, revela también una singularidad en nuestro país: en España se propagó un linaje del virus que apenas penetró en el resto de Europa

Covid-19 supercontagiadores. Mutaciones del SARS-Cov-2. Cepa B3a (Cepa más letal)
#sars_cov_2 #coronavirus #covid_19 #Supercontagiadores #CepaB3a http://
MILA MÉNDEZ
22/05/2020 11:57 H
Confiesan que están casi tan exhaustos como entregados a una investigación contrarreloj cuyos primeros resultados ya se están «viralizando». El genetista de la Facultade de Medicina de la USC e investigador del Instituto de Investigaciones Sanitarias (IDIS) Antonio Salas Ellacuriaga lidera un equipo de científicos que acumulan noches en vela estudiando el árbol genealógico del nuevo coronavirus y a sus huéspedes. Una tarea que también involucra al jefe de servicio de Pediatría del Complejo Hospitalario Universitario de Santiago (CHUS), Federico Martinón Torres, que es coordinador del grupo Genvip del IDIS, dentro del que se enmarca el proyecto internacional y multidisciplinar Gen-Covid. Después de semanas rastreando los linajes del SARS-CoV-2 han llegado varias conclusiones.
La primera involucra a los portadores. Entre un tercio y la mitad de todos los contagios registrados en el mundo se deben a los «supercontagiadores», personas con mayor sensibilidad a la transmisión del virus que provoca el covid-19 y de cuya existencia acaban de revelar las primeras pruebas. Para lograr su identificación, el equipo ha analizado casi 5.000 genomas del coronavirus de todo el mundo. En términos de código genético, aproximadamente 150 millones de letras que sometieron a análisis propios de genética evolutiva. Un «reto computacional» en palabras de Martinón Torres.
Hasta el momento, «la figura del ‘supercontagiador’ se había discutido en los medios y desde un punto de vista epidemiológico», apunta Salas. En algunos lugares han dado lugar a lo que los genetistas denominan técnicamente como efectos fundadores locales, que se han traducido en brotes epidémicos locales o nacionales.
«Dentro de estos casi 5.000 genomas analizamos, lo que vemos es que unas pocas docenas explican prácticamente la mitad de todos los casos. Esto quiere decir que el comportamiento epidemiológico-pandémico de esta infección se puede explicar por el papel primordial que algunos huéspedes tienen en la dispersión. No es que estas variantes, que estas cepas concretas sean más infectantes, sino que su combinación con unas características específicas del individuo explican esta rápida expansión en determinadas regiones y países», detalla Federico Martinón Torres, coordinador del grupo Genvip del IDIS.
Entre un tercio y la mitad de todos los contagios registrados en el mundo se deben a los «supercontagiadores»
El perfil del «supercontagiador»
«Se intuyen características en el perfil de estos 'supercontagiadores'. Personas con una mayor carga viral pero cuyos síntomas no son lo suficientemente llamativos o son asintomáticos. Otra es que sus tiempos de incubación sean muy largos, más de dos semanas, hasta 24 días como apuntan algunos trabajos iniciales. Durante ese tiempo son un magnífico contagiador. Otra característica es que tengan más secreciones respiratorias o más capacidad para proyectarlas. Por último, está su comportamiento, su grado de movilidad, especialmente, a lugares de riesgo», detalla el genetista Antonio Salas.
Partiendo del precepto de que el SARS-CoV-2 es un patógeno nuevo, el análisis del huésped, de su genética, es una de las especialidades de su grupo de investigación, centrado en enfermedades infecciosas. «En la neumococcemia, el rotavirus o el neumococo ya hemos demostrado en la literatura que el huésped juega un papel importante, no único, en todo esto. Lo que podríamos resumir en que no infecta siempre al que quiere, sino al que puede». Concretar el patrón de estas personas más susceptibles a transmitir el virus es uno de los campos en los que siguen trabajando.
El confinamiento frena las mutaciones
«Estamos viendo que la intervención humana funciona en la reducción de contagios, pero, además, desde el punto de vista genético se percibe en la variabilidad genómica. Cuando se frena la expansión también se frena la diversidad del patógeno. Su capacidad de mutar, algo que adquiere al generar nuevas copias de su ARN en los infectados. El pico del gran brote asiático fue el 29 de febrero. El 1 de marzo se observa un frenazo. Asia estaba confinada. Es a partir de ahí cuando determinados sublinajes se estancan. La genética habla de demografía,dice que el confinamiento ha cortado la diversificación del bicho», subraya Salas.
La genética habla de demografía, dice que el confinamiento ha cortado da diversificación del bicho
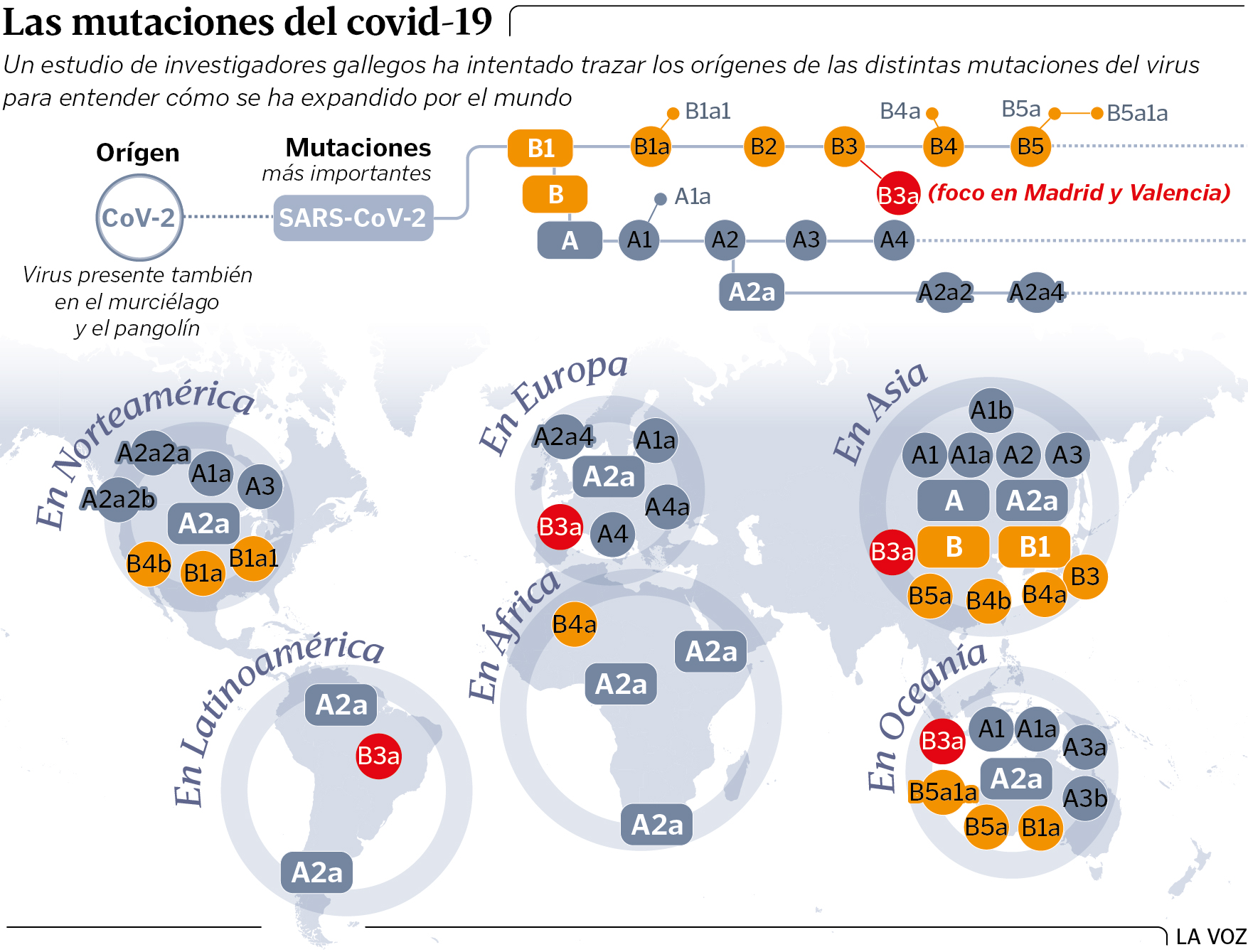
España: un caso único
La investigación arroja otro descubrimiento. Esta vez, en relación al genoma del coronavirus. «En nuestro país entraron las primeras cepas del virus que afectaron a casi toda Europa, pero a mayores, hay una que nos da un carácter distintivo, que no existe en el resto de países de nuestro entorno. Viene de China, no sé si de Wuhan», avanza el genetista del IDIS. Salas añade que «el escenario en España es un tanto particular en el contexto del continente».
Antes, hay que precisar que del SARS-CoV-2 parten dos grandes sublinajes de China. El A y B. El primero, el A2a, es el gran colonizador en el mundo fuera de Asia. El segundo, el B3a, sin embargo, es más exclusivo. Llega a nuestro país con una frecuencia que es excepcional en el resto del Europa. Hay unos pocos en Chile y otra rama en EE.UU. «Algunos linajes son muy llamativos en algunos sitios. En España, esta variación, si la observamos en el contexto de Europa, es especialmente llamativa», destaca Salas.
En el porqué de esta rareza, recalca, puede jugar un papel clave la suerte. «Hay un componente de azar importante en la expansión del virus. El genoma del que es portador una persona 'supercontagiadora' que llega a España se hace más fuerte en su zona porque, en un espacio de tiempo muy cortito, se transmite de forma muy rápida a mucha gente», explica el director de la investigación radicada en Santiago.
Eso sí, matiza: «No tenemos evidencias para mostrar que este linaje B3a sea más o menos agresivo. Por ahora no hemos detectado ninguna variación que presente unas características especiales que la hagan más peligrosa desde el punto de vista patogénico. Tampoco hemos tenido acceso a estos datos. La única asociación que hemos podido hacer es entre variante vírica y grupos de edad y sexo y no hemos detectado asociación concreta entre la primera y esas características biológicas».
Recibimos una cepa asiática que apenas entró en ningún otro país europeo
Noviembre del 2019: descartadas teorías conspirativas
Además, el estudio adelantado hoy jueves a la comunidad científica lleva parejas otras consideraciones relacionadas con la fecha probable del inicio de esta epidemia, la más importante del siglo, así como su posible origen que los investigadores sitúan en el mundo animal.
La variabilidad genética del coronavirus se corresponde a lo esperado por un proceso evolutivo natural. «Vemos una similitud superior al 96 % entre el genoma de los SARS-CoV-2 y el genoma del coronavirus de murciélago. Esto descarta las teorías conspiranoicas sobre el origen artificial del microorganismo y también apunta a ese origen animal, especialmente, dentro de la familia de los murciélagos», revela Martinón Torres.
«Usando simulaciones teóricas que se alimentan de la variabilidad genómica observada en el coronavirus, el trabajo sitúa de manera precisa el origen evolutivo más reciente de todos los linajes actuales no antes de noviembre de 2019» . En base a los datos recogidos y analizados, el equipo formado por Antonio Salas y Federico Martinón sugiere la posibilidad de que en la primera onda epidémica asiática pudieron existir muchos más casos que los reportados por las autoridades sanitarias.
Un virus «estable»
Por último, y no menos importante, está el carácter «estable» del virus. «Hemos encontrado que la similitud de estas casi 5.000 cepas es mayor 99,9% entre ellas, lo cual es una buena noticia desde el punto de vista de la variabilidad, del futuro desarrollo de vacunas e incluso del desarrollo epidemiólogico de la pandemia», avanza Federico Martinón.
En palabras del doctor Salas, «este es un paso fundamental para entender el proceso de dispersión del virus y nos será de gran utilidad para tratar de prever y prevenir futuros brotes y pandemias, ya sea de coronavirus u otros patógenos con potencial igualmente letal o incluso superior» .
«Teníamos claro que para entender lo que estaba ocurriendo en esta pandemia primero debíamos hacer una reconstrucción adecuada del proceso evolutivo que dio lugar al virus y sus distintas versiones actuales. Un árbol filogenético que relaciona todos los genomas de una manera precisa y que es el pilar fundamental sobre el que bascula casi todo lo demás», explica el docente de la Facultade de Medicina de la USC.
Según los autores, esta sería la primera vez que se utilizan los principios de máxima parsimonia para identificar las mutaciones concretas que dieron lugar a las distintas cepas del virus, «se trataba de aprovechar toda nuestra experiencia en el campo de la evolución genómica y llevarla al terreno del SARS-CoV-2», aporta Salas. Es decir, casar el campo de la infectómica con la genética de las poblaciones para una óptima filogenética del SARS-CoV-2.
Digerir toda la información en un período de tiempo tan breve no ha sido nada fácil. Según Federico Martinón, coordinador del grupo de investigación Genvip, «es la naturaleza coral de nuestro grupo y nuestra formación en el ámbito de la infectómica lo que nos ha permitido enfrentarnos a un proyecto de estas dimensiones». Distinas áreas del CHUS y del IDIS ha aportado su granito de arena. El Proyecto Gen Covid, continúa. «Esto solo es el principio», informa Martinón. Para seguir avanzado es vital que contar con más financiación.

El caso del supercontagiador que transmitió el coronavirus a 24 personas en un viaje de autobús
En enero de este año, un solo hombre contagió a varios pasajeros en un viaje de una hora y cuarenta minutos, gracias al sistema de ventilación del vehículo y debido a que nadie llevaba mascarilla
En enero de este año, un solo hombre contagió a varios pasajeros en un viaje de una hora y cuarenta minutos, gracias al sistema de ventilación del vehículo y debido a que nadie llevaba mascarilla
September 1, 2020
Community Outbreak Investigation of SARS-CoV-2 Transmission Among Bus Riders in Eastern China

No hay comentarios:
Publicar un comentario